In part 1 of this tutorial we looked at how we can solve the read alignment problem using Burrows-Wheeler transform. In this post, we will continue exploring this algorithm and see how we can do read alignment with mismatches allowed.
The read alignment problem is: You are given millions of small pieces of DNA sequence, called reads and one large DNA sequence called the reference sequence (DNA sequences contain only 4 letters A, C, G, T representing the four bases). You have to find how many of those reads matches the reference sequence and at what positions. In simplified form, you have many shorter strings to be matched against a single very large string. We already saw how we can do this exact matching. But often the reference DNA string and the small reads come from different persons. And there can be differences between them because of the mutation. On average, there’s a difference of 1 in 1000 characters between the DNA sequence of two people. So we need to match the reads, but we can allow that few of the characters are not an exact match. This problem can arise in other applications too. In short, we have one large string and many small strings to be matched against that one string and we can allow a fixed number of mismatched characters. How can we do it fast?
We will be extending the concepts from the previous post. When doing exact matching, we reduced the band size based on the next character. But now we will create 4 different bands (because we have 4 characters in DNA sequences) and call BWA on all of them. We will also keep a counter of mismatches left. Out of those 4 calls, 1 will be of exact match and other 3 of a mismatch (with mismatches left counter reduced by 1). We’ll keep on calling recursively until mismatches left is reduced to 0 and we find a mismatch, or we completely parsed the read string, the case of a match.
This pseudo-code should make it clear:
BWA(band_start, band_end, mismatch_left, read, index):
if(index == 0):
return band_start, band_end
if(band_start == band_end):
return None
ans = []
next_character = read[index-1]
for each symbol s:
rank_top = Rank(s, band_start)
rank_bottom = Rank(s, band_end)
if(s == next_character): # exact match
ans += BWA(bands[s] + rank_top,
bands[s] + rank_bottom,
mismatch_left, read, index-1)
else if (mismatch_left > 0): # mismatch
ans += BWA(bands[s] + rank_top,
bands[s] + rank_bottom,
mismatch_left-1, read, index-1)
return ans
Where the bands store the starting indices of the band for each character as in the previous post. The initial call will be (where N is the length of reference sequence):
BWA(1, N+1, mismatches_allowed, read, len(read))
Let’s follow through an example: CGATGCACCGGT.
First, it’s BWT:
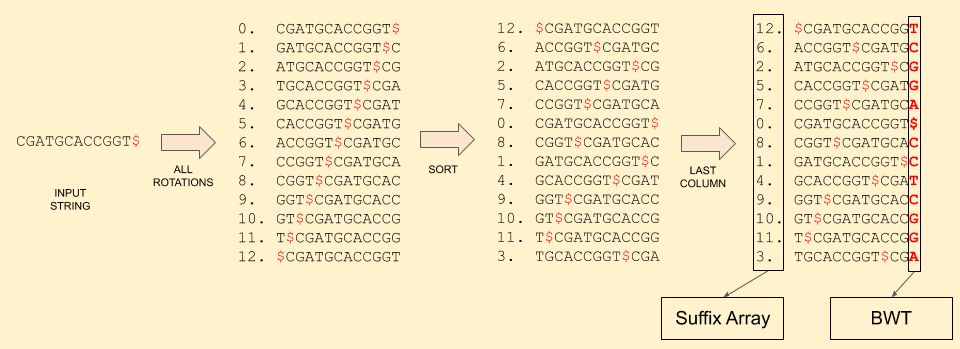
Reference sequence: C G A T G C A C C G G T $
Burrows-Wheeler: T C G G A $ C C T C G G A
Suffix Array: 12 6 2 5 7 0 8 1 4 9 10 11 3
Let our read to be matched be: CGA. With 1 mismatch allowed.
We start with A. And start 4 BWA calls, 1 with the exact match of A and 3 others with a mismatch. In each of the 4 calls, we also save the number of mismatches left. Since, we already used a mismatch for C, G and T, mismatch_left counter becomes 0. While for A, it’s still 1.
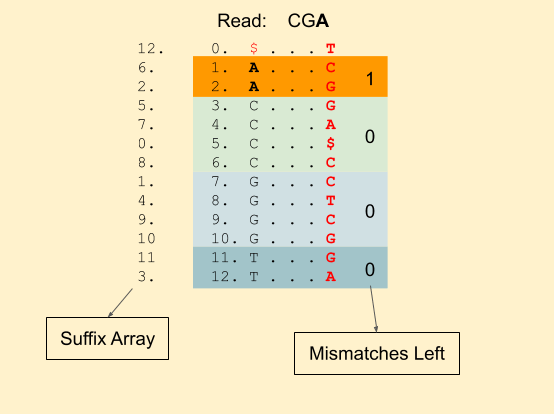
The 4 bands we have are (1, 3), (3, 7), (7, 11), (11, 13). Next, we have a G. Now for each of these 4 bands, we will create 4 other bands. Our recursive calls will look like this (For clarity, I have removed the read from the calls, we have band_start, band_end, mismatches_left and index in order). It also shows the further recursive calls.
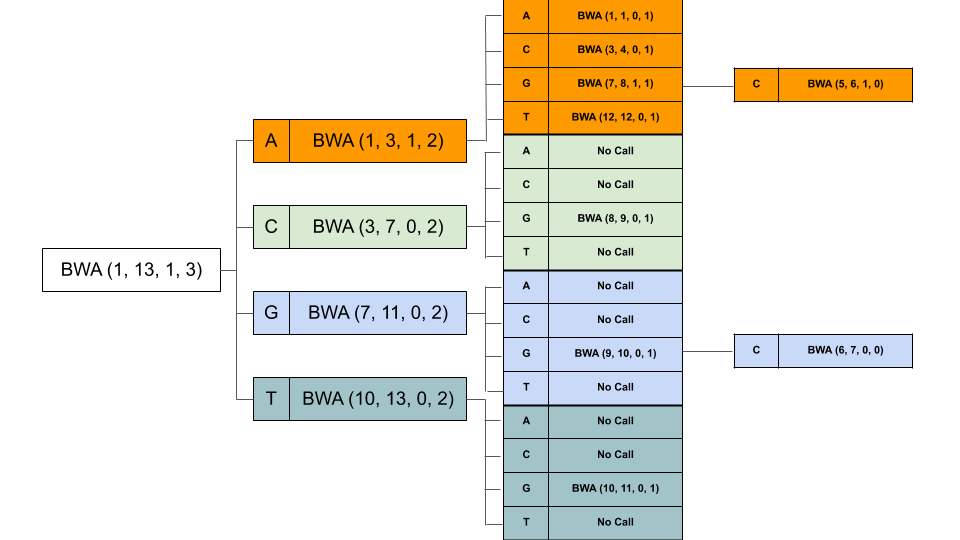
At last, all but 2 calls returned None, with bands (5, 6) and (6, 7). That means we found matches at band (5, 7), i.e. indices 5 and 6. From suffix array at these indices, we get 0 and 8. That means, there is a match at position 0 and position 8 in the original reference string. Note, at index 0, its an exact match, while at index 8 we found CGG (instead of CGA), there is 1 mismatch (G instead of A).
Reference sequence: C G A T G C A C C G G T $ Index: 0 1 2 3 4 5 6 7 8 9 . . .
That’s it for this post. Burrows-Wheeler Alignment is a great algorithm. There have been many other improvements on this basic algorithm. One is how to handle insertions and deletions (indels). Can you come up with it yourself? Give it a try!